November 5, 2013
Stanton A. Glantz, PhD
FDA process for approving “Substantially Equivalent” tobacco products keeps the public in the dark
Under the 2009 Family Smoking and Prevention Tobacco Control Act (FSPTCA) there are two ways in which the FDA can approve a new tobacco product to be marketed: (1) a tobacco company can submit a “new product” application, or (2) it can submit a “substantial equivalence” application to the FDA making the case that the new product is “substantially equivalent” to a product already on the market as of February 15, 2007.
To date there have been over 4,300 substantial equivalence applications (and no new product applications), so the scientific standards that the FDA uses to determine that a new product is “substantially equivalent” are very important in terms of determining the public health impact of approving the new product. It is, therefore, particularly important that the public know precisely on what grounds such decisions are being made.
Amidst great fanfare, FDA announced on June 25, 2013 that for the first time since FSPTCA gave it authority in 2009 to regulate tobacco products, FDA approved the marketing of two new tobacco products (Lorillard Newport Non-Menthol Gold Box 100s and Newport Non-Menthol Gold Box) and denied the marketing of four others through the substantial equivalence (SE) pathway. This process grandfathered tobacco products existing as of February 15, 2007, but required FDA approval for new products, permitting manufacturers tosubmit a substantial equivalence report demonstrating that its new tobacco product is substantially equivalent to a product already on the market. In its official press release, Mitch Zeller, director of the FDA’s Center for Tobacco Products, stated: “The FDA is committed to making science-based decisions on all product applications and providing the agency’s scientific rationale behind its actions to ensure the most transparent and efficient process possible for all involved parties.”
This sounds great, but the reality is that as of November 2013 the FDA is still keeping the public in the dark about its criteria for making SE decisions.
Here is the tale of our efforts to get this information:
- In its June 25, 2013 SE approval letters [press release] for Lorillard's new Newport Non-Menthol Gold Box 100s and Newport Non-Menthol Gold Box the FDA stated that Lorillard “agreed” to make health information related to the new tobacco products available upon request by any member of the public. As noted above, this information is critical for judging whether the FDA applied a reasonable scientific standard to making its decision.
- On July 8, 2013, we asked Lorillard for that information.
- On July 17, 2013, Lorillard responded that if we were interested in more than the very perfunctory health information report Lorillard reported on its website, they “recommended” that we submit a FOIA request to FDA.
- On August 2, 2013, we submitted a FOIA request to FDA.
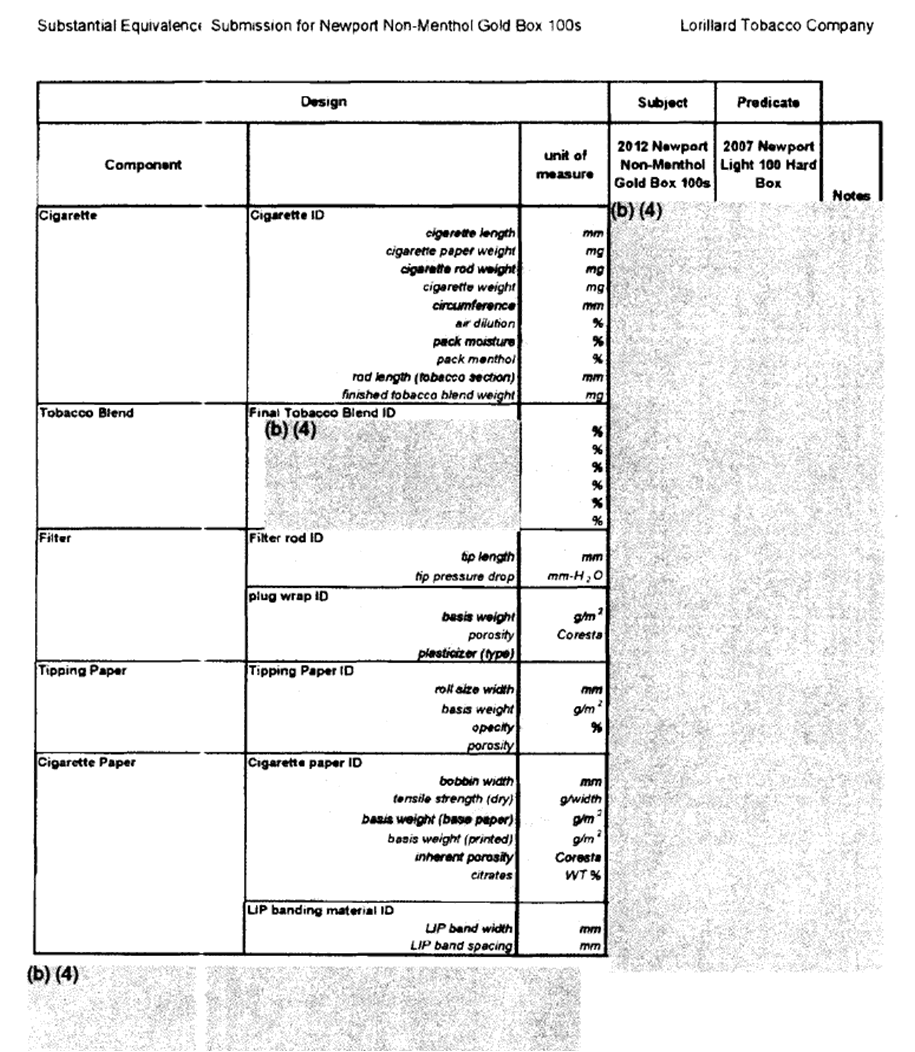
- On August 13, 2013, the FDA responded with 197 pages of documentation, including Lorillard's applications and the FDA's analysis for Lorillard's SE submissions that redacted every scientifically important detail on the ground of “trade secret”. [link] (See example page below.) The FDA even redacted readily available public information such as cigarette length that is readily available to the public.
- On August 9, 2013, in consultation with other interested parties, we wrote a detailed letter to Mitch Zeller expressing concern about the complete lack of transparency in the process, outlining the back-and-forth referral process between the FDA, Lorillard, and back again and laid out a legal requirement for the FDA being more forthcoming about the specifics of the specific information used to make SE decisions.
- On October 23, 2013, Zeller responded with a letter that ignored the substance of our letter and simply restated the FDA’s commitment to “transparency.” He did not provide any new scientific information.
In particular, Zeller ignored the four specific questions at the end of our letter:
- In light of the strong disclosure requirements in Section 910(a)(4)(A) [of the FSPTCA], why do your [SE] letters say that Lorillard “agreed” to provide information to the public, suggesting that some sort of negotiating with the FDA was permitted or encouraged, as well as saying that Lorillard “may wish to consider providing” A or B, or alternatively, they “may provide” A, B, C, D, and E?
- Why did FDA use optional, rather than mandatory, language in its letters to Lorillard?
- Why did FDA invite Lorillard to make a statement, saying that “Lorillard does not have or know of any research or data regarding adverse health effects….” when it is not delineated by the law?
- Did FDA require Lorillard to submit the information required by Section 910(B)(1), and did it rely on this information to make its SE determination? If not, why not, and what did FDA rely on?
Because of the FDA’s indiscriminate redactions and its refusal to answer the four questions above, it is impossible for the public to tell what standards and processes it uses to determine “substantial equivalence” and impossible to understand the “agency’s scientific rationale behind its actions.” This information is critically important to the individual members of the public so they can make appropriate decisions about whether or not to purchase and use the approved tobacco products, and for the scientific community to investigate the harmfulness of these products and to comment on the FDA’s actions.
On January 21, 2009, President Obama signed his Memorandum on Transparency and Open Government which affirmed his administration’s commitment to “creating an unprecedented level of openness in Government” in order to “strengthen our democracy and promote efficiency and effectiveness in Government.” Noting that “information maintained by the Federal Government is a national asset,” the President called on executive departments and agencies to “put information about their operations and decisions online and readily available to the public.”
In June 2009, FDA Commissioner Margaret Hamburg launched FDA’s three-phase “Transparency Initiative” that included launching the Phase I web-based resource called “FDA Basics” in January 2010, releasing the “Phase II Transparency Report” in May 2010 that focused on disclosing certain information about FDA-regulated products and firms, and releasing the “Phase II Transparency Report” that focused on increasing the transparency of FDA operations and decision-making.
As President Obama said in his memorandum on Transparency, “public engagement enhances the Government’s effectiveness and improves the quality of its decisions. Knowledge is widely dispersed in society, and public officials benefit from having access to that dispersed knowledge.”
It is time for the FDA to disclose information that is complete and useful to the public, and that demonstrates the scientific rationale behind is decisions on SE applications. Federal law and regulations authorize and require it to do so.
Two pages from the FDA’s response to us on how they determined that Lorillard’s SE application warranted approval. The “(b)(4)” redactions (referring to the trade secret exemption in FOIA).

Lauren Lempert helped me prepare this blog post.
Comments
FDA needs to be transparent about SE applications
Stan,
I share your concerns about the FDA's lack of transparency for evaluating SE applications, especially since the agency has only approved 9 SE applications (including 2 for manufactured cigarettes, and 7 for RYO tobacco and papers). The FDA also has rejected 13 SE applications so far (but hasn't revealed any info on those products), and has a huge backlog of ;4,000 SE applications.
Since manufactured cigarettes, RYO cigarette papers, RYO tobacco and smokeless tobacco products are all vastly different products, the FDA should have developed different criteria to evaluate SE applications for each of those four different types of products, and should make those criteria public.
Please note that unless the FDA exempts newly deemed tobacco products (i.e. e-cigs, little cigars, large cigars, smoking tobacco, shisha/hookah tobacco, dissolvables, nicotine skin creams) from Section 905(j) and Section 910 regulations for SE applications, the deeming regulation would ban ALL newly deemed tobacco products (that are currently on the market) from being introduced into the US market pending FDA approval of an SE application, which poses a conundrum for FDA (and exposes the incompetence of negotiators, sponsors and advocates of the TCA).
But assuming the FDA exempts newly deemed tobacco from Section 905(j) and Section 910 requirements (to avoid entirely justifiable lawsuits), the FDA should also develop different criteria for evaluating SE applications for each of the different types of newly deemed tobacco products.
Last week, the FDA issued a Brief Summary of “Not Substantially Equivalent” Determinations hinting at why <1% of SE tobacco product applications have been approved or rejected, and explaining (as I've done many times) why the FDA deeming regulation would ban all e-cigs currently on the market (since none are Substantially Equivalent to a product on the market before February 15, 2007) .
http://www.fda.gov/downloads/TobaccoProducts/Labeling/MarketingandAdvert...
According to that document, the FDA can reject any SE application simply because the agency claims to have "inadequate information" on the product.
Bill Godshall
Executive Director
Smokefree Pennsylvania
1926 Monongahela Avenue
Pittsburgh, PA 15218
412-351-5880
[email protected]
The FDA should reject SE applications that have inadequate info
That's a no brainer.
Add new comment